Содержание:
- 1 Гипохондроплазия — причины, симптомы, лечение
- 1.1 Ахондроплазия
- 1.2 Краткая информация
- 1.3 Что такое ахондроплазия?
- 1.4 Признаки и симптомы
- 1.5 Причины и факторы риска
- 1.6 Затронутые группы населения
- 1.7 Похожие на ахондроплазию расстройства
- 1.8 Диагностика
- 1.9 Стандартные методы лечения
- 1.10 Причины развития и лечение гипохондроплазии
- 1.11 Факторы, влияющие на развитие болезни
- 1.12 Симптоматика описываемой патологии
- 1.13 Диагностика
- 1.14 Можно ли вылечить недуг?
- 1.15 Гипохондроплазия
- 1.16 Общие сведения
- 1.17 Причины гипохондроплазии
- 1.18 Симптомы гипохондроплазии
- 1.19 Диагностика
- 1.20 Лечение гипохондроплазии
- 1.21 Профилактика
- 1.22 Гипохондроплазия
- 1.23 Гипохондроплазия
- 1.24 Гипохондроплазия
- 1.25 Причины гипохондроплазии
- 1.26 Симптомы гипохондроплазии
- 1.27 Диагностика гипохондроплазии
- 1.28 Лечение и прогноз гипохондроплазии
- 1.29 Гипохондроплазия: клиническая картина, симптомы, лечение, прогноз
- 1.30 Патогенез заболевания
- 1.31 Клиническая картина гипохондродисплазии
- 1.32 Диагностика
- 1.33 Проявления и лечение гипохондроплазии
- 1.34 Этиологические факторы
- 1.35 Симптоматическая картина
- 1.36 Диагностирование
- 1.37 Терапевтический подход
- 1.38 Ахондроплазия
- 1.39 Содержание
- 1.39.1 Определение и общие сведения [ править ]
- 1.39.2 Этиология и патогенез [ править ]
- 1.39.3 Клинические проявления [ править ]
- 1.39.4 Ахондроплазия: Диагностика [ править ]
- 1.39.5 Дифференциальный диагноз [ править ]
- 1.39.6 Ахондроплазия: Лечение [ править ]
- 1.39.7 Профилактика [ править ]
- 1.39.8 Прочее [ править ]
- 1.40 Гипохондроплазия
- 1.41 Содержание
- 1.42 Диагностика
- 1.43 Профилактика
- 1.44 Хондродисплазии: классификация, нарушения, диагностика, лечение
- 1.45 Классификация
- 1.46 Распространенность
- 1.47 Молекулярные нарушения
Гипохондроплазия — причины, симптомы, лечение
Ахондроплазия
Краткая информация
Ахондроплазия (также называемая: диафизарная аплазия, болезнь Парро-Мари, врожденная хондродистрофия) — это хрящевая болезнь, которая является одной из основных причин непропорциональной карликовости: у больных пациентов верхние и нижние конечности короче, чем обычно, а туловище — нормальное.
Причины ахондроплазии генетические: мутация гена FGFR3, расположенного на хромосоме 4, вызывает ее возникновение.
Помимо характерных особенностей короткого роста и отсутствия пропорциональности между конечностями и туловищем, ахондроплазия является причиной других клинических признаков, включая: коротких пальцев, вальгусной деформации колен или стоп, большой головы и видного лба.
В настоящее время не существует специальных методов лечения ахондроплазии. Поэтому ахондроплазия является неизлечимым заболеванием.
Что такое ахондроплазия?
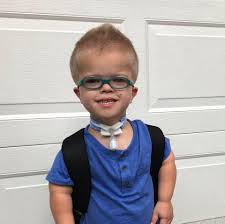
Ахондроплазия — это редкое генетическое (наследственное) заболевание костей, которое встречается у одного из 15 000–35 000 живорожденных.
Ахондроплазия — наиболее распространенный тип карликовости, при котором руки и ноги ребенка коротки пропорционально длине тела. Кроме того, голова часто бывает большой, а туловище нормального размера.
Средний рост взрослых мужчин с ахондроплазией составляет около 133 см. Средний рост взрослых женщин с ахондроплазией составляет около 125 см.
Признаки и симптомы

Это редкое генетическое заболевание характеризуется отличительными признаками:
- низкий рост (обычно ниже 5 футов [см. фото]);
- необычно большая голова (макроцефалия) с выраженным лбом (лобная выпуклость) и плоским (вдавленным) носовым мостиком;
- короткие руки и ноги;
- выпуклый живот и ягодицы (из-за внутренней кривой позвоночника);
- короткие пальцы рук, которые принимают трехконечное положение во время разгибания.
Признаки в детском возрасте.
Дети, рожденные с ахондроплазией, обычно имеют «куполообразный» (сводчатый) череп и очень широкий лоб. В небольшой пропорции происходит чрезмерное накопление жидкости вокруг мозга (гидроцефалия). Для ахондроплазии в младенчестве также характерен низкий мышечный тонус (гипотония).
Причины и факторы риска
Ахондроплазия возникает в результате специфических изменений (мутаций) гена, известного как рецептор 3 фактора роста фибробластов (FGFR3).
Для большинства пациентов нет очевидного семейного анамнеза этого состояния. Повышенный возраст отца (пожилой отцовский возраст) может быть фактором, способствующим возникновению спорадической ахондроплазии.
Реже семейные случаи ахондроплазии следуют по аутосомно-доминантному типу наследования. Доминантные генетические расстройства возникают, когда необходима только одна копия аномального гена, чтобы вызвать конкретное расстройство.
Аномальный ген может быть унаследован от любого из родителей или может быть результатом мутации (изменения) гена у пострадавшего человека. Риск передачи ненормального гена от пораженного родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Ахондроплазия поражает мужчин и женщин в равных количествах. Это расстройство появляется у развивающегося плода и является одной из наиболее распространенных форм скелетной дисплазии, вызывающей карликовость. Предполагаемая частота ахондроплазии варьировала от 1 на 15 000 до 1 на 35 000 рождений.
Похожие на ахондроплазию расстройства
Симптомы следующих расстройств могут быть сходными с таковыми при ахондроплазии. Сравнения могут быть полезны для дифференциальной диагностики:
Гипохондроплазия — это генетическое заболевание, характеризующееся небольшим ростом и непропорционально короткими руками, ногами, кистями и ступнями (т. е. карликовостью коротких конечностей). У тех, кто страдает этим расстройством, низкий рост часто не распознается до раннего или среднего возраста или, в некоторых случаях, до зрелого возраста. У пострадавших также может развиться изгиб ног в раннем детстве, который часто самопроизвольно улучшается с возрастом. В некоторых случаях могут присутствовать дополнительные аномалии, такие как необычно большая голова (макроцефалия), относительно выступающий лоб, ограниченное разгибание и вращение локтей и/или другие физические отклонения.
Кроме того, примерно в 10 процентах случаев может наблюдаться легкая умственная отсталость. В некоторых случаях гипохондроплазия, по-видимому, возникает случайно по неизвестным причинам (спорадически) без очевидного семейного анамнеза. В других случаях это семейное расстройство с аутосомно-доминантным наследованием. Как отмечалось выше (см. «Причины»), гипохондроплазия и ахондроплазия могут быть результатом различных мутаций одного и того же гена (т.е. FGFR3).
Ахондроплазию можно отличить от других форм карликовости коротких конечностей посредством тщательного клинического обследования, рентгенологического исследования и/или дополнительных диагностических методов.
Диагностика
Клинические и рентгенологические особенности ахондроплазии хорошо охарактеризованы. Те, у кого типичные результаты, как правило, не нуждаются в молекулярно-генетическом тестировании для подтверждения диагноза.
Когда клинические особенности вызывают подозрение у новорожденного, рентгенологические данные могут помочь подтвердить диагноз. Однако, если есть неопределенность, идентификация генетического варианта гена FGFR3 с помощью молекулярно-генетического тестирования может быть использована для установления диагноза.
Стандартные методы лечения
В настоящее время нет способа предотвратить или лечить ахондроплазию, поскольку в большинстве случаев возникают новые неожиданные мутации. Лечение гормоном роста существенно не влияет на рост человека с ахондроплазией. В некоторых очень специализированных случаях могут быть рассмотрены удлиняющие ноги операции.
Обнаружение аномалий кости, особенно в области спины, важно для предотвращения затрудненного дыхания и болей в ногах или потери функции. Кифоз (или горбун) может потребоваться хирургическая коррекция, если он не исчезает, когда ребенок начинает ходить. Хирургия также может помочь сгибанию ног. Ушные инфекции, возникают вследствие болезни необходимо лечить немедленно, чтобы избежать потери слуха. Проблемы с зубами, возможно, должны быть решены ортодонтом (стоматологом, имеющим специальную подготовку по выравниванию зубов).
У большинства людей с ахондроплазией ожидаемая продолжительность жизни будет нормальной или близкой к нормальной. Однако существует повышенный риск преждевременной смерти, связанный не только с внезапной неожиданной смертью в младенчестве, но и, по-видимому, с сердечно-сосудистыми осложнениями в зрелом возрасте.
В целом средняя продолжительность жизни примерно на 10 лет меньше, чем у населения в целом. Недавно завершенное исследование подтверждает, что самые высокие стандартные показатели смертности наблюдаются в возрасте до 4 лет. Однако, кроме того, это многоцентровое исследование смертности показывает, что произошло резкое снижение смертности, в том числе внезапных неожиданных смертей, у маленьких детей с ахондроплазией, скорее всего, вторично по отношению к признанию их особых рисков и агрессивной оценки и вмешательства, связанных с краниоцервикальным соединением.
Причины развития и лечение гипохондроплазии
Гипохондроплазия — это наследственная болезнь, вызывающая нарушения в процессе формирования костных структур. Такие поражения приводят к карликовости. Гипохондроплазия встречается довольно редко, но по сравнению с другими подобными патологиями протекает в легких формах, хотя поражает как мужчин, так и женщин в равной степени.

Факторы, влияющие на развитие болезни
Причины этой болезни следующие:
- Торможение роста хондроцитов и фибробластов при развитии ребенка.
- Развитие карликовости костей из-за замены в белках, формирующих кости, вещества аспарагина лизином.
- Множественность подмен азотистых оснований в гене. По этой причине изменяется состав протеина.
 В результате происходит нарушение формирование костных структур и хрящевой ткани, которое приводит к уменьшению размеров костей. Мутации такого рода передаются от родителей детям при помощи аутосомно-доминантного механизма, но по большей части они спонтанны, поэтому выявляются только у детей, а родители и родственники ребенка могут иметь нормальную костную структуру. Хотя болезнь может развиться с большой степенью вероятности в семьях, где один из родителей болен этим заболеванием.
В результате происходит нарушение формирование костных структур и хрящевой ткани, которое приводит к уменьшению размеров костей. Мутации такого рода передаются от родителей детям при помощи аутосомно-доминантного механизма, но по большей части они спонтанны, поэтому выявляются только у детей, а родители и родственники ребенка могут иметь нормальную костную структуру. Хотя болезнь может развиться с большой степенью вероятности в семьях, где один из родителей болен этим заболеванием.
Симптоматика описываемой патологии
Признаки болезни начинают проявляться в возрасте 3–4 лет. До этого периода ребенок не имеет никаких отклонений при родах, хорошо набирает вес, развивается, как и все другие дети. Затем начинается отставание от сверстников в росте, у ребенка увеличиваются кисти и ступни, хотя у него остаются короткие конечности.
Развивается практически незаметное ограничение движений в локтях, появляются симптомы поясничного лордоза. У таких детей нет никакого отставания от сверстников в умственной сфере. В глазах окружающих ребенок выглядит как невысокий, коренастый человек.
При обследовании ребят старше 4 лет врачи фиксируют характерные патологии в виде:
- укороченных конечностей;
- увеличенного размера стопы и кисти;
- нарушений подвижности локтей или тазобедренного сустава.
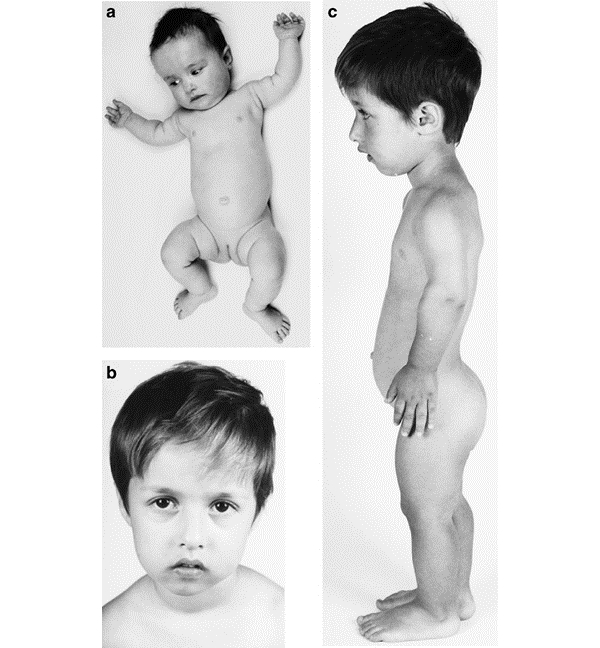 Гипохондроплазия не изменяет форму лица или черепа больного, хотя иногда у таких пациентов врачи фиксируют брахицефалию. У больных редко фиксируются контрактуры на суставах локтя или тазобедренном суставе, но нет искривления костей на бедрах.
Гипохондроплазия не изменяет форму лица или черепа больного, хотя иногда у таких пациентов врачи фиксируют брахицефалию. У больных редко фиксируются контрактуры на суставах локтя или тазобедренном суставе, но нет искривления костей на бедрах.
У половины больных развивается лордоз на поясничном отделе позвоночного столба. Часть пациентов жалуются на боль в ногах при передвижении или подъеме по лестнице.
Все эти нарушения чаще всего появляются у ребят, отец которых в момент зачатия был старше 40–45 лет.
Как выглядит пациент с таким заболеванием, что это такое, можно увидеть на фото, размещенных в медицинских справочниках.
Диагностика
Анализ данных и постановку диагноза осуществляют педиатр и ортопед на основе рентгенологического обследование пациента, проведения генетического и молекулярного исследования.
Все эти нарушения фиксируются при наружном обследовании, а затем подтверждаются рентгеном костных структур. Выявляется сужение спинномозгового канала на позвоночном столбе, вогнутость контуров задней поверхности на поясничных позвонках.
Рентген дает возможность исследовать уплотнение костей на плечах и бедрах ребенка, небольшое удлинение большой берцовой кости, позволяет выявить уплощение вертлюжной впадины. При этом диагностируется:
- укорочение локтевой косточки;
- наличие квадратных эпифизов костей на коленном суставе.
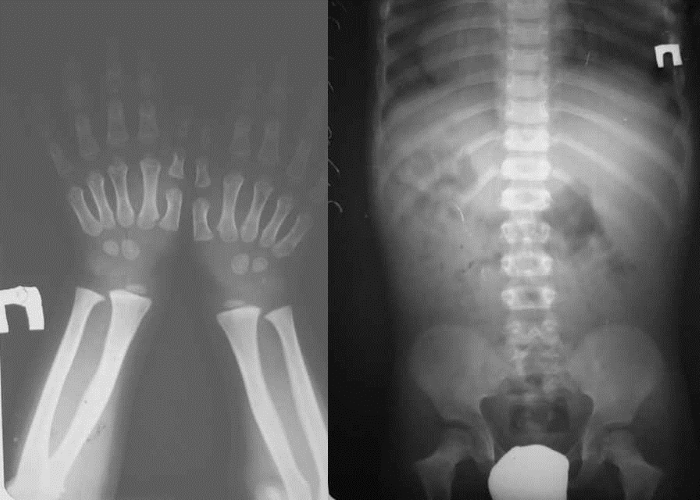
Генетическая экспертиза позволяет выявить изменения, произошедшие из-за мутации гена, отвечающего за формирование костей. Для этого исследуется 13-й ген, где чаще всего врачи находят нарушения.
Делается дифференциальный диагноз, позволяющий отличить описываемую болезнь от подобных ей заболеваний. Такая диагностика базируется на рентгеновском обследовании и генетических исследованиях.
Можно ли вылечить недуг?
Лечение описываемой болезни практически не проводится из-за того, что у человека лордоз и контрактуры на локтях выражены довольно слабо, нет умственного отставания от других людей. Прогноз жизни человека с описываемой болезнью благоприятен.
Иногда, когда человек уже становится взрослым, ему может потребоваться операция из-за сдавливания нервных корешков поясничной зоны спинного мозга. Болезненность возникает из-за лордоза и нарушения строения позвонков.
Удлинить кости можно такими методами, как:
- гормональное лечение;
- применение аппарата Елизарова и т. д.

Гормональные препараты довольно дороги, причем они не позволяют быстро увеличить длину костей. Чаще всего больной вынужден использовать их на протяжении всей жизни.
Использование методики Елизарова предполагает ломку костей пациента, удлинение костных структур механическим методом.
Группа инвалидности при этом заболевании не назначается, хотя есть инструкции Министерства здравоохранения, в которых прописаны случаи «укорочения конечностей», когда больному можно дать инвалидность.
Гипохондроплазия
Гипохондроплазия – наследственное заболевание, относящееся к группе хондродисплазий, причина которого заключается в нарушении формирования хрящей и некоторых типов костей, что приводит к карликовости. Симптомами этой патологии являются укороченные конечности и пальцы, увеличенные относительные размеры кистей и стоп, небольшое ограничение движений в локтевом суставе и поясничный лордоз. Диагностика производится на основании осмотра пациента, данных рентгенологических исследований, молекулярно-генетического анализа. Лечения гипохондроплазии не существует, однако серьезных осложнений, значительно ухудшающих качество жизни пациента (кроме карликовости), также не наблюдается.
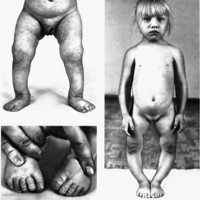
Общие сведения
Гипохондроплазия – генетическая патология, которая во многом сходна с родственными ей хондродисплазиями – например, ахондроплазией и танатоформной дисплазией. Впервые это состояние было описано в 1913 году Равенной, который выявил наследственную природу этой формы карликовости и ее отличия от ахондроплазии. Встречаемость гипохондроплазии в настоящий момент точно неизвестна – некоторые врачи-генетики указывают цифры 1:100000, однако с этим согласны далеко не все исследователи.
Основное затруднение в вычислении встречаемости этого заболевания обусловлено тем, что более чем в 80% случаев оно возникает спонтанно по причине мутаций de novo. Остальная доля случаев гипохондроплазии (менее 20%) передается по аутосомно-доминантному механизму. Однако точно установлено, что данная патология встречается намного реже ахондроплазии (1:20000), хоть и имеет более легкое течение. Заболевание с равной степенью вероятности поражает как мужчин, так и женщин.
Причины гипохондроплазии
Большое сходство проявлений гипохондроплазии с ахондроплазией обусловлено тем, что причиной данных заболеваний являются мутации одного и того же гена — FGFR3, расположенного на 4-й хромосоме. Он кодирует последовательность фактора роста фибробластов-3, представляющего собой трансмембранный рецептор тирозинкиназы. В норме он несколько тормозит рост фибробластов и хондроцитов в ростовых пластинках эндохондральных костей, тем самым контролируя правильное развитие костной ткани. При мутациях гена FGFR3 полученный фактор роста фибробластов-3 имеет дефект и не может полноценно выполнять свои функции – в зависимости от характера генетического дефекта это может привести к танатоформной дисплазии, ахондроплазии или гипохондроплазии.
Выявлено несколько десятков мутаций гена FGFR3, результатом которых является развитие карликовости по типу гипохондроплазии. Наиболее распространенным является дефект, возникающий по причине замены азотистого основания (аденина или гуанина) в положении 1620 тринадцатого экзона гена. В результате в полученном от такого гена белке в положении 540 происходит замена аспараганина на лизин, что значительно изменяет тирозинкиназную чувствительность рецептора.
Кроме того, описано еще множество других замен азотистых оснований в гене и, как следствие, аминокислот в полученном протеине, которые приводят к развитию симптомов гипохондроплазии. Однако имеются указания, что у некоторых больных с клинической картиной этого заболевания при генетическом исследовании не было выявлено дефектов FGFR3. Это может говорить о том, что в развитии гипохондроплазии могут участвовать и другие гены.
Все мутации вышеуказанного гена наследуются по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев они являются спонтанными и не выявляются у родителей или родственников больного. Подмечено, что у многих пациентов с гипохондроплазией возраст отцов превышает средний, что может указывать на герминативную природу появления мутаций гена FGFR3.
Симптомы гипохондроплазии
Как правило, в первые годы жизни ребенка гипохондроплазия ничем себя не проявляет – при рождении не наблюдается никаких отклонений от нормы, набор массы и психофизическое развитие происходят нормально. Первые признаки отставания в росте регистрируются в возрасте 3-4-х лет, становится заметной небольшая диспропорциональность тела (короткие руки и ноги, увеличенные ступни и кисти). Во многих случаях окружающие даже не сразу выявляют какие-либо необычные пропорции у больных – они просто выглядят невысокими коренастыми людьми. Форма черепа и черты лица при гипохондроплазии зачастую без особенностей, иногда может выявляться небольшая брахицефалия.
У больных иногда возникают незначительные сгибательные контрактуры локтевых суставов и очень редко – тазобедренных. Вальгусное искривление бедренных костей при гипохондроплазии не наблюдается, возможны аналогичные деформации голени. Примерно в половине случаев у больных возникает лордоз поясничного отдела позвоночника. Так как все эти проявления похожи на симптомы ахондроплазии, только выражены намного слабее, долгое время считалось, что это одна и та же патология, только отдельные исследователи относили ее к самостоятельной нозологической единице. Лишь современные исследования в области генетики однозначно доказали справедливость такого выделения гипохондроплазии.
Диагностика
Выявление гипохондроплазии производится на основании комплекса медицинских мер: консультации педиатра и ортопеда, рентгенологических исследований, молекулярно-генетического анализа. При осмотре больных старше 3-4-х лет обнаруживают характерные для патологии изменения – укороченные конечности, увеличенный относительный размер стоп и кистей, нарушения подвижности в локтевом и иногда в тазобедренном суставе, поясничный лордоз. Если гипохондроплазия имеет наследственный характер, то такие проявления всегда имеются у одного из родителей, так как это заболевание является аутосомно-доминантным. При здоровых родителях косвенным признаком, указывающим на эту патологию, может быть возраст отца на момент зачатия более 40-45 лет.
Намного больше информации о гипохондроплазии дают рентгенологические исследования костей, суставов и позвоночника. При обследовании последнего выявляют сужение спинномозгового канала в каудальном направлении, вогнутые контуры задней поверхности поясничных позвонков. Также рентгенологически наблюдается укорочение и уплотнение бедренных и плечевых костей, незначительное удлинение большеберцовой кости, эпифизы костей квадратной формы в области коленных суставов, уплощение вертлужной впадины. Примерно в двух третях случаев гипохондроплазии также диагностируется укорочение локтевой кости.
Генетическая диагностика гипохондроплазии включает в себя прямое секвенирование последовательности гена FGFR3 или отдельных его экзонов для выявления мутаций. Чаще всего производят исследование 13-го экзона гена, так как именно там располагаются дефекты в большинстве случаев заболевания. Возможна пренатальная диагностика посредством амниоцентеза или биопсии ворсин хориона.
Дифференциальный диагноз следует проводить с ахондроплазией и другими состояниями, которые сопровождаются укорочением конечностей. Различают эти патологии между собой при помощи рентгенологических и генетических методов исследования.
Лечение гипохондроплазии
Специфического лечения гипохондроплазии не существует, карликовость остается у человека на всю жизнь. Однако ряд других проявлений по типу контрактуры суставов и лордоза поясничного отдела позвоночника выражены умеренно. Поэтому прогноз гипохондроплазии относительно жизни и ее качества в большинстве случаев благоприятный. Лишь в некоторых случаях взрослым больным может потребоваться хирургическое вмешательство из-за сдавления поясничного отдела спинного мозга или его корешков (радикулит), которые возникают по причине лордоза и нарушения строения позвонков.
Профилактика
Профилактика заболевания сводится к пренатальной генетической диагностике, особенно это необходимо делать семейным парам, где один из родителей страдает от гипохондроплазии. Учитывая аутосомно-доминантный характер наследования этой патологии, вероятность рождения больного ребенка при здоровом втором родителе составляет 50%.
Гипохондроплазия
OMIM 146000
Наша команда профессионалов ответит на ваши вопросы
Гипохондроплазия – хондродистрофия, форма карликовости с короткими конечностями, имеющая сходство с ахондроплазией. Эти виды заболеваний отличаются клинически и по рентгенологическим исследованиям. В отличие от ахондроплазии при гипохондроплазии голова и лицо нормальных размеров, строение таза нормальное. Пальцы короткие, но их расположение нормальное, а не в форме трезубца, как при ахондроплазии. Нет искривления большеберцовой кости. Нет неврологических нарушений. Малая берцовая кость не укорочена. У больных широкие кисти и стопы. Спинномозговой канал сужается в каудальном направлении. Расстояние между корнями дужек поясничных позвонков в каудальном направлении не суживается, как при ахондроплазии, но и не расширяется, как в норме, а остается примерно одинаковым. У больных рентгенологически выявляются вогнутые контуры задней поверхности поясничных позвонков, укорочение и утолщение плечевых и бедренных костей, «квадратная» форма эпифизов коленных суставов, укорочение локтевой кости в области лучезапястного сустава. Симптомы заболевания проявляются в 3-4 года.
Гипохондроплазия наследуется по аутосомно-доминантному типу. Примерно 80% случаев развиваются в результате возникновения мутации de novo.
Гипохондроплазия обусловлена мутациями в гене рецептора фактора роста фибробластов-3 FGFR3 (MIM 134934), картированном в районе 4р16.3. Ген FGFR3 состоит из 19 экзонов и имеет длину 16.5 т.п.н. В 40-60% случаев к заболеванию приводит замена в 540 положении аспарагина на лизин (Asn540Lys) в проксимальном тирозинкиназном домене. К данной замене аминокислоты приводят замены нуклеотида в 1620 положении (С1620А или С1620G) в экзоне 13 гена FGFR3. Кроме того к фенотипу гипохондроплазии приводят мутации: Asn540Thr, Asn540Ser, Ile538Val, Lys650Asn (в результате замены нуклеотида G1950T или G1950C), Lys650Gln, Lys652Gln и ряд других мутаций, располагающихся в основном в 5, 7, 10, 13 и 15 экзонах гена FGFR3.
Однако, гипохондроплазия является генетически гетерогенным заболеванием. Описаны семьи, в которых сцепление с геном FGFR3 исключено.
В Центре Молекулярной Генетики проводится поиск частых мутаций, приводящих к развитию гипохондроплазии, расположенных в экзоне 13 гена FGFR3: Asn540Lys (замена нуклеотидов С1620А, С1620G), Asn540Ser (замена нуклеотидов А1619G). Кроме того проводится поиск частых мутаций, приводящих к развитию ахондроплазии, расположенных в экзоне 10 гена FGFR3: Gly380Arg (замена нуклеотидов G1138A, G1138C), Gly375Cys (замена нуклеотидов G1123T). Для проведения поиска мутаций в гене FGFR3 необходимо предоставить свежую кровь, взятую в пробирку с консервантом ЭДТА.
Анализ является наиболее полным и включает поиск мутаций, характерных не только для гипохондроплазии, но и для ахондроплазии.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Гипохондроплазия

Гипохондроплазия – наследственное заболевание, относящееся к группе хондродисплазий, причина которого заключается в нарушении формирования хрящей и некоторых типов костей, что приводит к карликовости. Симптомами этой патологии являются укороченные конечности и пальцы, увеличенные относительные размеры кистей и стоп, небольшое ограничение движений в локтевом суставе и поясничный лордоз. Диагностика производится на основании осмотра пациента, данных рентгенологических исследований, молекулярно-генетического анализа. Лечения гипохондроплазии не существует, однако серьезных осложнений, значительно ухудшающих качество жизни пациента (кроме карликовости), также не наблюдается.
Гипохондроплазия

Гипохондроплазия – генетическая патология, которая во многом сходна с родственными ей хондродисплазиями – например, ахондроплазией и танатоформной дисплазией. Впервые это состояние было описано в 1913 году Равенной, который выявил наследственную природу этой формы карликовости и ее отличия от ахондроплазии. Встречаемость гипохондроплазии в настоящий момент точно неизвестна – некоторые врачи-генетики указывают цифры 1:100000, однако с этим согласны далеко не все исследователи. Основное затруднение в вычислении встречаемости этого заболевания обусловлено тем, что более чем в 80% случаев оно возникает спонтанно по причине мутаций de novo. Остальная доля случаев гипохондроплазии (менее 20%) передается по аутосомно-доминантному механизму. Однако точно установлено, что данная патология встречается намного реже ахондроплазии (1:20000), хоть и имеет более легкое течение. Заболевание с равной степенью вероятности поражает как мужчин, так и женщин.
Причины гипохондроплазии
Большое сходство проявлений гипохондроплазии с ахондроплазией обусловлено тем, что причиной данных заболеваний являются мутации одного и того же гена — FGFR3, расположенного на 4-й хромосоме. Он кодирует последовательность фактора роста фибробластов-3, представляющего собой трансмембранный рецептор тирозинкиназы. В норме он несколько тормозит рост фибробластов и хондроцитов в ростовых пластинках эндохондральных костей, тем самым контролируя правильное развитие костной ткани. При мутациях гена FGFR3 полученный фактор роста фибробластов-3 имеет дефект и не может полноценно выполнять свои функции – в зависимости от характера генетического дефекта это может привести к танатоформной дисплазии, ахондроплазии или гипохондроплазии.
Выявлено несколько десятков мутаций гена FGFR3, результатом которых является развитие карликовости по типу гипохондроплазии. Наиболее распространенным является дефект, возникающий по причине замены азотистого основания (аденина или гуанина) в положении 1620 тринадцатого экзона гена. В результате в полученном от такого гена белке в положении 540 происходит замена аспараганина на лизин, что значительно изменяет тирозинкиназную чувствительность рецептора. Кроме того, описано еще множество других замен азотистых оснований в гене и, как следствие, аминокислот в полученном протеине, которые приводят к развитию симптомов гипохондроплазии. Однако имеются указания, что у некоторых больных с клинической картиной этого заболевания при генетическом исследовании не было выявлено дефектов FGFR3. Это может говорить о том, что в развитии гипохондроплазии могут участвовать и другие гены. Все мутации вышеуказанного гена наследуются по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев они являются спонтанными и не выявляются у родителей или родственников больного. Подмечено, что у многих пациентов с гипохондроплазией возраст отцов превышает средний, что может указывать на герминативную природу появления мутаций гена FGFR3.
Симптомы гипохондроплазии
Как правило, в первые годы жизни ребенка гипохондроплазия ничем себя не проявляет – при рождении не наблюдается никаких отклонений от нормы, набор массы и психофизическое развитие происходят нормально. Первые признаки отставания в росте регистрируются в возрасте 3-4-х лет, становится заметной небольшая диспропорциональность тела (короткие руки и ноги, увеличенные ступни и кисти). Однако отклонений в умственной сфере при гипохондроплазии практически никогда не наблюдаются. Во многих случаях окружающие даже не сразу выявляют какие-либо необычные пропорции у больных – они просто выглядят невысокими коренастыми людьми. Форма черепа и черты лица при гипохондроплазии зачастую без особенностей, иногда может выявляться небольшая брахицефалия.
У больных иногда возникают незначительные сгибательные контрактуры локтевых суставов и очень редко – тазобедренных. Вальгусное искривление бедренных костей при гипохондроплазии не наблюдается, возможны аналогичные деформации голени. Примерно в половине случаев у больных возникает лордоз поясничного отдела позвоночника. Так как все эти проявления похожи на симптомы ахондроплазии, только выражены намного слабее, долгое время считалось, что это одна и та же патология, только отдельные исследователи относили ее к самостоятельной нозологической единице. Лишь современные исследования в области генетики однозначно доказали справедливость такого выделения гипохондроплазии.
Диагностика гипохондроплазии
Выявление гипохондроплазии производится на основании комплекса медицинских мер: консультации педиатра и ортопеда, рентгенологических исследований, молекулярно-генетического анализа. При осмотре больных старше 3-4-х лет обнаруживают характерные для патологии изменения – укороченные конечности, увеличенный относительный размер стоп и кистей, нарушения подвижности в локтевом и иногда в тазобедренном суставе, поясничный лордоз. Если гипохондроплазия имеет наследственный характер, то такие проявления всегда имеются у одного из родителей, так как это заболевание является аутосомно-доминантным. При здоровых родителях косвенным признаком, указывающим на эту патологию, может быть возраст отца на момент зачатия более 40-45 лет.
Намного больше информации о гипохондроплазии дают рентгенологические исследования костей, суставов и позвоночника. При обследовании последнего выявляют сужение спинномозгового канала в каудальном направлении, вогнутые контуры задней поверхности поясничных позвонков. Также рентгенологически наблюдается укорочение и уплотнение бедренных и плечевых костей, незначительное удлинение большеберцовой кости, эпифизы костей квадратной формы в области коленных суставов, уплощение вертлужной впадины. Примерно в двух третях случаев гипохондроплазии также диагностируется укорочение локтевой кости.
Генетическая диагностика гипохондроплазии включает в себя прямое секвенирование последовательности гена FGFR3 или отдельных его экзонов для выявления мутаций. Чаще всего производят исследование 13-го экзона гена, так как именно там располагаются дефекты в большинстве случаев заболевания. Возможна пренатальная диагностика посредством амниоцентеза или биопсии ворсин хориона. Дифференциальный диагноз следует проводить с ахондроплазией и другими состояниями, которые сопровождаются укорочением конечностей. Различают эти патологии между собой при помощи рентгенологических и генетических методов исследования.
Лечение и прогноз гипохондроплазии
Специфического лечения гипохондроплазии не существует, карликовость остается у человека на всю жизнь. Однако в отличие от ахондроплазии это заболевание практически никогда не сопровождается умственной неполноценностью, ряд других проявлений по типу контрактуры суставов и лордоза поясничного отдела позвоночника также выражены весьма незначительно. Поэтому прогноз гипохондроплазии относительно жизни и ее качества в большинстве случаев благоприятный. Лишь в некоторых случаях взрослым больным может потребоваться хирургическое вмешательство из-за сдавления поясничного отдела спинного мозга или его корешков (радикулит), которые возникают по причине лордоза и нарушения строения позвонков. Профилактика заболевания сводится к пренатальной генетической диагностике, особенно это необходимо делать семейным парам, где один из родителей страдает от гипохондроплазии. Учитывая аутосомно-доминантный характер наследования этой патологии, вероятность рождения больного ребенка при здоровом втором родителе составляет 50%.
Гипохондроплазия: клиническая картина, симптомы, лечение, прогноз
Гипохондросплазия – врожденное состояние, при котором идет нарушение формирования как суставов, так и хрящевой ткани. в итоге это приводит к изменению формы и размера сочленений, существенно уменьшая их размер.
Такое состояние вызывает состояние карликовости.
Патогенез заболевания
 Это генетический тип заболевания, который относится к врожденным состояниям. Гипохондроплазия во многом сходна с другими состояниями вроде ахондроплазии, танатоморфной дисплазии и так далее.
Это генетический тип заболевания, который относится к врожденным состояниям. Гипохондроплазия во многом сходна с другими состояниями вроде ахондроплазии, танатоморфной дисплазии и так далее.
Подобное состояние описано было впервые в 1913 году. Статистика по патологии отличается от источника к источнику, но обычно указывается, что данное состояние выявляют в 1 случае из 100000.
Такая разница в данных объясняется простым фактором – более 80% больных получили патологию спонтанно из-за мутаций типа de novo. И только 20% пациентов получили болезнь по наследственному пути. Точно известно, что эта патология встречается реже ахондроплазии и протекает, как правило, в более легкой форме.
Клиническая картина гипохондродисплазии
 Обычно в первые годы жизни заболевание себя не выдает никакими признаками. Отклонений от норм при рождении не выявляют, набор массы, психофизическое развитие ребенка идет в нормальном темпе.
Обычно в первые годы жизни заболевание себя не выдает никакими признаками. Отклонений от норм при рождении не выявляют, набор массы, психофизическое развитие ребенка идет в нормальном темпе.
Первые признаки в отношении отставания роста могут проявиться, начиная с 3-4 лет. В таком случае становится заметной некоторая диспропорциональность тела в виде укорочения ног, рук, увеличения кистей и ступней.
В умственной сфере заболевание почти никогда не проявляется. В большинстве случаев окружение ребенка не всегда выявляет даже физические отклонения в развитии. Обычно такие дети кажутся просто невысокими, в некоторой степени коренастыми людьми. Черты лица и форма черепа обычно не меняются, но в ряде случаев все же фиксируется брахицефалия.
У больных также могут возникать небольшие контрактуры сгибательного типа в области локтей и чуть реже – тазобедренных суставов. Могут иногда проявляться в области голени проявления вальгусной деформации.
Но современные исследования показали, что гипохондроплазия является отдельной нозологической единицей.
Причины выделяются в целом две. В большинстве случаев болезнь провоцируют спонтанные мутации в генах. Только пятая часть больных получает патологию наследственным способом. Пол, экология, иные факторы, как правило, никакой роли в проявлении состояния не играют.
 Симптомы у болезни проявляются не особенно выражено. В первые 3-4 года у ребенка каких-либо отклонений или нарушений не определяют. Но начиная с указанного возраста могут проявиться:
Симптомы у болезни проявляются не особенно выражено. В первые 3-4 года у ребенка каких-либо отклонений или нарушений не определяют. Но начиная с указанного возраста могут проявиться:
- Некоторое укорочение верхних и нижних конечностей;
- Увеличение размера кистей и ступней;
- Вальгусная деформация голеней;
- Замедление роста;
- Приобретение типичной коренастой внешности;
- Невыраженные сгибательные контрактуры;
- Лордоз в области поясницы;
- Редко – брахиоцефалия.
На фото зоны роста при гипохондродисплазии

Диагностика
Диагностировать подобное состояние во время беременности не удается, так как состояние не проявляет себя визуально даже после рождения в первые годы жизни. Крайне редко в период вынашивания ребенка проводится амниоцентез, а также биопсия ворсин хориона, при исследовании которых могут быть выявлены мутации соответствующих генов. Но такие исследования в большинстве случаев не проводят.
Только после того, как начались изменения, происходит замедление роста, врачи могут заподозрить болезнь. В таком случае проводится тщательный осмотр ребенка, после чего назначается прохождение:
 Лечения у такого состояния нет. Гипохондроплазия провоцирует развитие карликовости, которая остается до конца жизни.
Лечения у такого состояния нет. Гипохондроплазия провоцирует развитие карликовости, которая остается до конца жизни.
Но в отличии от схожих патологий, данное состояние не провоцирует существенных ограничений в образе жизни, а симптоматика проявляется невыраженно.
Умственная сторона развития не затрагивается, из-за чего врачи лечением данной патологии просто не занимаются.
Из-за лордоза и контрактуры, которые также носят низкую интенсивность проявления, могут быть прописаны курсы физиотерапии и ЛФК.
Учитывая все вышесказанное, можно сделать вывод, что гипохондроплазия – это врожденное заболевание, которое не оказывает в контексте здоровья на ребенка негативного воздействия. Потому прогнозы у таких больных благоприятные, отзывы после оперативного лечения положительные.
Единственный фактор, который следует учитывать – это необходимость адаптации и укрепления психоэмоциональной стороны развития, чтобы больной мог нормально функционировать в социуме без психологических травм и комплексов, спровоцированных недостаточно высоким ростом.
Отзывы о лечении гипохондроплазии у ребенка:
Проявления и лечение гипохондроплазии
Содержание:
 Гипохондроплазия относится к генетическим заболеваниям, сходным с родственными ей хондродисплазиями (ахондроплазией и танатоформной дисплазией). В первый раз болезнь описал в 1913 г Равенна, выявивший наследственный генез такой разновидности карликовости и ее различия с ахондроплазией.
Гипохондроплазия относится к генетическим заболеваниям, сходным с родственными ей хондродисплазиями (ахондроплазией и танатоформной дисплазией). В первый раз болезнь описал в 1913 г Равенна, выявивший наследственный генез такой разновидности карликовости и ее различия с ахондроплазией.
Распространенность на современном этапе до конца не определена, часть ученых отмечают частоту 1:100000. Главная трудность в исчислении распространенности этой патологии заключена в спонтанном возникновении почти в 80% случаев по причине мутирований de novo. Остальная часть имеет аутосомно-доминантный путь наследования. Болезнь с одинаковой степенью поражает как мужской, так и женский пол.
Этиологические факторы
Большое схожесть клиники с ахондроплазией обуславливается мутированием одного гена FGFR3, который располагается на IV хромосоме, который кодирует последовательность фактора роста фибробластных клеток-3. В нормальных условиях последний немного тормозит рост этих и хондральных клеток в ростковых пластинах эндохондральных костей, чем и контролирует нормальное развитие остеоткани. В случае мутирования этот фермент приобретает дефект и не способен правильно функционировать. В соответствии с характером генетического дефекта патпроцесс может свести к танатоформной дисплазии, гипохондроплазии либо ахондроплазии.
Симптоматическая картина
Обычно на первых годах жизни малыша гипохондроплазия протекает бессимптомно, т.е. при отсутствии каких-либо отклонений в развитии. Первоначальные проявления отставания в росте отмечаются на третьем-четвертом году, тогда уже визуализируется легкое нарушение пропорциональности туловища (укороченные ручки и ножки, увеличенные стопы и кисти). Но отставания в умственной сфере практически отсутствуют. В основном окружающие не сразу замечают какие-то необычные пропорции у заболевшего ребенка – вид у него невысокого коренастого человечка. Череп и черты лица часто также без каких-то собенных характеристик, в некоторых случаях отмечается незначительная брахицефалия.
Иногда развиваются небольшие сгибательные контрактуры локтей и крайне редко – ТБС. Вальгусная деформация феморальных костей отсутствует, возможно такое деформирование в голенной части. Примерно в 50% развивается лордоз люмбальной части позвоночного столба. Поскольку перечисленные признаки похожи на картину при ахондроплазии, лишь менее выражены, длительный период полагали это одной болезнью, а некоторые исследователи относили ее к самостоятельному заболеванию. ТИ только на свлременном этапе в области генетики получили доказательства развития гипохондроплазии.
Диагностирование
Диагностика осуществляется на основе физикального осмотра педиатра и ортопеда, ренгенисследований,генетического анализа на молекулярном уровне. При осмотре пациентов после трехлетнего возраста выявляются типичные для болезни трансформации. В случае наследственной природы всегда отмечаются сходные проявления у какого-то родителя.
Наиболее информативным может стать рентгенологическое обследование костного аппарата, суставных сочленений позвоночных сегментов, в этом случае определяется стеноз спинального канала в каудальную сторону, вогнутые границы задней плоскости люмбальных позвонков. Дополнительно рентгенологически отмечается укорочение и уплотнение бедра и плечевой кости, небольшое удлинение большой берцовой кости, эпифизарные части имеют прямоугольную форму в зоне коленного сочленения, уменьшение глубины вертлюжных впадин. Примерно в 60% случаев укорачивается локтевая кость.
Генетическое исследование подразумевает определение последовательности ДНК и РНК гена FGFR3 либо некоторых его экзонов с целью обнаружения мутирования. Зачастую выполняют оценку XIII экзона гена, поскольку там в основном. Существует пренатальное диагностирование с помощью амниоцентеза либо биопсии хориональных ворсинок. Дифдиагноз выполняется с ахондроплазией и иными патсостояниями с укорочением рук и ног. Отличия устанавливают на основе генного обследования и ренгенисследования.
Терапевтический подход
 Специфической терапии сегодня не существует, карликовость сохраняется у пациента. Но в отличие от ахондроплазии такая патология никогда не протекает с умственной отсталостью, а суставная контрактура и люмбальный лордоз не особо выражен. Поэтому жизненный прогноз в большей части считается благоприятным. Только в определенных случаях взрослым пациентам назначается оперативный способ лечения по причине компрессии люмбального участка спинномозговой ткани либо нейрокорешков, развивающегося из-за лордоза и аномальной структуры позвонковых сегментов. Профилактические мероприятия основываются на пренатальном генетическом диагностировании (особенно, если существует риск семейной заболеваемости). Учитывая аутосомно-доминантный путь передачи патпроцесса, риск рождения ребенка с аналогичным заболеванием при здоровом другом родителе в пределах 50%.
Специфической терапии сегодня не существует, карликовость сохраняется у пациента. Но в отличие от ахондроплазии такая патология никогда не протекает с умственной отсталостью, а суставная контрактура и люмбальный лордоз не особо выражен. Поэтому жизненный прогноз в большей части считается благоприятным. Только в определенных случаях взрослым пациентам назначается оперативный способ лечения по причине компрессии люмбального участка спинномозговой ткани либо нейрокорешков, развивающегося из-за лордоза и аномальной структуры позвонковых сегментов. Профилактические мероприятия основываются на пренатальном генетическом диагностировании (особенно, если существует риск семейной заболеваемости). Учитывая аутосомно-доминантный путь передачи патпроцесса, риск рождения ребенка с аналогичным заболеванием при здоровом другом родителе в пределах 50%.
Ахондроплазия
Рубрика МКБ-10: Q77.4
Содержание
Определение и общие сведения [ править ]
Ахондроплазия — системное поражение скелета, врожденная болезнь, характеризуемая нарушением энхондрального остеогенеза и проявляющаяся карликовостью, укорочением конечностей при обычной длине туловища, деформацией нижних конечностей и позвоночника и относительной макроцефалией.
Заболеваемость ахондроплазией составляет 1,3 на 100 000 населения.
Этиология и патогенез [ править ]
Ахондроплазия развивается в результате впервые возникшей или унаследованной доминантной мутации. В результате беспорядочного расположения клеток росткового хряща нарушается процесс энхондрального окостенения, и рост костей в длину замедляется.
Тип наследования — аутосомно-доминантный, 80% случаев обусловлены новыми мутациями. Заболевание вызвано мутацией гена рецептора-3 фактора роста фибробластов, картированного в локусе 4p16.3.
Клинические проявления [ править ]
Многие дети гибнут внутриутробно. Уже при рождении у ребенка можно обнаружить макроцефалию и микромелию. Увеличение мозговой части черепа и выпуклость лба обусловлены аномалией развития хрящевой основы черепа. Желудочки мозга значительно увеличены, но давление внутри желудочков, как правило, не выходит за пределы нормы. Вследствие выраженного нарушения развития костей основания черепа лицо больных приобретает характерные черты: выдающиеся вперед лобные кости, седловидный нос («выскобленное ложечкой лицо»), прогнатия. Поражается весь скелет, но особенно выражены изменения в проксимальных отделах конечностей. Укорочение конечностей вначале носит ризомелический характер, т.е. больше укорочены проксимальные сегменты (плечо и бедро). Это объясняется тем, что плечевая и бедренная кости в норме растут особенно интенсивно, и задержка роста в этих костях особенно заметна. Вследствие извращенного и замедленного эпифизарного роста при ненарушенном периостальном росте все трубчатые кости утолщены, изогнуты, бугристы из-за выступания апофизов; эпифизы деформированы. Это ведет к варусным и вальгусным деформациям, которые прогрессируют при ранней нагрузке на нижние конечности. Прибавляются и вторичные деформации — разболтанность связок коленного сустава, плосковальгусная деформация стопы вследствие неправильности оси конечностей.
В первые месяцы жизни в области конечностей заметны кожные складки и жировые подушки. Связки коленных суставов всегда перерастянуты. Дети, больные ахондроплазией, отстают в физическом развитии, поздно начинают держать головку (после 3-4 мес) и сидеть (после 8-9 мес), а ходить начинают к 1,5-2 годам. Уже на первом году жизни у многих больных появляется кифоз в поясничном отделе позвоночника. По мере роста ребенка укорочение конечностей становится более заметным.
Компрессионный синдром проявляется в трех формах: компрессия нервных корешков на уровне конского хвоста; компрессия спинного мозга на уровне грудо-поясничного отдела позвоночника и компрессия спинного мозга на уровне верхних шейных позвонков (вследствие стеноза большого затылочного отверстия). Поясничный гиперлордоз усиливает сдавление элементов спинного мозга и может превышать 90°. Стеноз большого затылочного отверстия может вызвать компрессию спинного мозга на уровне верхних шейных позвонков с парестезией по ходу плечевого сплетения. Подвижность па уровне атланто-окципитального сочленения может отсутствовать в результате его недоразвития.
Взрослые достигают высоты 131 ± 5,6 см (мужчины) и 124 ± 5,9 см (у женщин).
Ахондроплазия: Диагностика [ править ]
Патогномоничный рентгенологический признак — сужение расстояния между ножками дуг поясничных позвонков в каудальном направлении. Кроме того, крылья подвздошной кости развернуты, укорочены, имеют прямоугольную форму; крыши вертлужных впадин горизонтальные. Поперечный размер входа в малый таз значительно превышает его глубину. Метафизарные отделы длинных костей утолщены, бокаловидно расширены, эпифизы погружены в них по типу шарнира; диафизы укорочены, выглядят истонченными по сравнению с массивными и утолщенными метафизами и вертельными областями. Суставные поверхности деформированы, неконгруэнтны. Типичны деформации коленного и лучезапястного суставов. Малоберцовая кость относительно удлинена, принимает участие в образовании сустава. Щель сустава расширена, эпифизы имеют неправильную форму.
Дифференциальный диагноз [ править ]
Дифференциальный диагноз включает гипохондроплазию, летальную карликовость (тип I и II) и синдром.
Ахондроплазия: Лечение [ править ]
Консервативное лечение в раннем возрасте направлено на профилактику деформаций нижних конечностей, укрепление мышц конечностей, спины, живота.
Хирургические вмешательства при ахондроплазии направлены на коррекцию варусных деформаций нижних конечностей и низкорослости.
Варусные деформации голеней корригируют подмыщелковыми и надлодыжечными остеотомиями. Удлинение нижних конечностей для ребенка представляет сложный комплекс отрицательных эмоций, вызванных практически постоянной, хотя и не интенсивной, болью в месте остеотомий и проведения спиц и стержней, неудобствами при ходьбе и самообслуживании. Редкие дети до 10-летнего возраста осознают необходимость проводимого удлинения. Именно поэтому возраст 10 лет считают оптимальным сроком для начала лечения.
Предпочтительней, чтобы рост пациента при начале удлинения составлял минимум 100 см. Это дает перспективу после окончания всех этапов удлинения достигнуть роста 150 см.
Предоперационное обследование пациентов включает исследование состояния внутренних органов, тщательное неврологическое обследование для исключения гидроцефалии и стеноза позвоночного канала в поясничном отделе. Стеноз позвоночного отверстия в поясничных позвонках (а в отдельных наблюдениях и стеноз большого затылочного отверстия) может стать причиной тяжелых осложнений, поэтому в предоперационном периоде показаны КТ, МРТ позвоночника, а иногда и миелография.
На конечностях оценивают состояние кровообращения и функциональное состояние нервных стволов. Существует множество различных схем удлинения сегментов. Опыт диктует, что к этой проблеме необходим дифференцированный подход.
Осложнения при удлинении сегментов довольно типичны. Это неврологические или сосудистые расстройства, возникшие в процессе операции или дистракции, воспалительные процессы, развитие контрактур смежных суставов. Лечат их по известным схемам. В случае неврологических расстройств, связанных со стенозом спинномозгового канала, показана декомпрессивная ламинэктомия с последующей стабилизацией металлоконструкцией.
Восстановительное лечение после удлинения направлено на борьбу с контрактурами суставов, избыточным весом больного. Важно выработать правильный стереотип ходьбы в условиях новой биомеханической ситуации. После окончания лечения все усилия ортопеда и пациента должны быть направлены на профилактику ранних артрозов.
Профилактика [ править ]
Прочее [ править ]
Определение и общие сведения
Гипохондроплазия характеризуется наличием непропорциональной низкорослости, мягкого поясничного лордоза и ограниченным разгибанием локтевых суставов.
Распространенность оценивается на уровне около 1 в 33000. Гипохондроплазия передается по аутосомно-доминантному типу.
Этиология и патогенез
Гипохондроплазия вызывается мутацией гена рецептора фактора роста фибробластов 3 (FGFR3; 4p16.3), однако локализация мутаций иная, чем при ахондроплазии, а именно в проксимальном домене тирозинкиназы (TK1) FGFR3-N540K (Asn540Lys) и Ile538Val.
Клинические проявления обычно становятся очевидными в детстве. Дисморфизм лица, ортопедические пороки развития и неврологические проблемы не присущи данной патологии. Сообщается о случаях интеллектуального дефицитома и искривления нижних конечностей.
Типичная рентгенологическая картина включает умеренное сужение межножковых расстояний, укороченные трубчатые кости с дистальным удлинением малоберцовой кости, короткая и широкая шейка бедра.
Гипохондроплазия близко напоминает ахондроплазию (также вызвана мутацией гена FGFR3), но имеет менее выраженную низкорослость и диспропорцию скелетна, а также более мягкую рентгенологическую картину.
Лечение только симптоматическое.
Рост у взрослых пациентов колеблется между 132 и 147 см, а продолжительность жизни в норме.
Синдром тяжелая ахондроплазия-задержка развития-акантокератодермия (acanthosis nigricans)
Синдром тяжелая ахондроплазия-задержка развития-акантокератодермия был описано у четырех не связанных между собой пациентов.
Сообщалось также об аномалиях центральной нервной системы, судорогах и потере слуха, а также в некоторых случаях деформации ключицы, бедренной кости, голени и малоберцовой кости.
Синдром вызван замещением Lys650Met в киназном домене рецептора фактора роста 3 фибробластов, кодируемого геном FGFR3 (4p16.3).
Гипохондроплазия

Содержание
Значительное сходство симптомов гипохондроплазии с ахондроплазией обусловлено тем, что оба эти заболевания возникают в результате мутации одного и того же гена — FGFR3, расположенного на 4 хромосоме, отвечающей за кодировку последовательности фактора роста фибробластов-3, который представляет собой трансмембранный рецептор тирозинкиназы. В норме он незначительно ингибирует рост фибробластов и хондроцитов в ростовых пластинках эндохондральных костей, что позволяет ему контроливать правильное развитие костной ткани. При мутациях гена FGFR3 фактор роста фибробластов-3 является неполноценным, что не позволяет ему правильно выполнять его функции и иногда приводит к развитию танатоформной дисплазии, ахондроплазии или гипохондроплазии.
Выявлено более двадцати мутаций гена FGFR3, в результате которых происходит развитие карликовости по типу гипохондроплазии. Самым распространенным типом дефекта является дефект, возникающий в результате замены азотистого основания в положении 1620 тринадцатого экзона гена. В результате в полученном от такого гена белке в положении 540 происходит замена аспараганина на лизин, что полностью изменяет тирозинкиназную чувствительность рецептора. Помимо этого, описано достаточно много других замен азотистых оснований в гене и, как следствие, аминокислот в полученном протеине, которые вызывают развитие гипохондроплазии. Однако некоторые сведения указывают на то, что у больных с клинической картиной этого заболевания при генетическом исследовании не было выявлено дефектов FGFR3. Это указывает на то, что в развитии гипохондроплазии могут участвовать и другие гены. Все мутации вышеуказанного гена наследуются по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев они являются спонтанными и не выявляются у родителей или родственников больного. Подмечено, что у многих лиц с гипохондроплазией возраст отцов превышает средний, что может указывать на герминативный генез мутаций гена FGFR3.
В большинстве случаев в первые годы жизни ребенка заболевание никак не проявляет себя. При рождении у таких детей не обнаруживается никаких отклонений от нормы, так как набор массы тела и психофизическое развитие происходят нормально. Первые признаки отставания в росте появляются у ребенка в возрасте 3 или 4 лет. В этом возрасте у малыша становится заметной незначительная диспропорциональность тела. Нарушение интеллектуального развития при гипохондроплазии практически никогда не наблюдаются. Во многих случаях окружающие даже не сразу выявляют какие-либо необычные пропорции у больных, вследствие чего они просто выглядят невысокими коренастыми людьми. Форма черепа и черты лица при гипохондроплазии зачастую без особенностей, иногда может определяться небольшая брахицефалия.
У больных в редких случаях могут возникать незначительные сгибательные контрактуры локтевых суставов и очень редко – тазобедренных. У больных не выявляется вальгусного искривления бедренных костей при гипохондроплазии, но при этом возможны аналогичные деформации голени. Примерно в половине случаев у пациентов возникает лордоз поясничного отдела позвоночника. В связи с тем, что все эти явления похожи на симптомы ахондроплазии, только менее выражены, долгое время считалось, что это одна и та же патология, и только отдельные исследователи относили ее к самостоятельной нозологической единице, что было подтверждено генетическими исследованиями.
Диагностика
Диагностика гипохондроплазии осуществляется на основании комплекса медицинских мероприятий. Для постановки диагноза больному может потребоваться проведение рентгенологических исследований и молекулярно-генетического анализа.
На данный момент не разработано специфическое лечения гипохондроплазии, вследствие этого карликовость остается у человека на всю жизнь. Однако в отличие от ахондроплазии данная патология практически никогда не сопровождается умственной неполноценностью. В терапевтических целях такие больные могут получать витаминно-минеральные комплексы. В редких случаях взрослым больным может потребоваться хирургическое вмешательство из-за сдавления поясничного отдела спинного мозга или его корешков, которые возникают по причине лордоза и нарушения строения позвонков.
Профилактика
Профилактика гипохондроплазии сводится к пренатальной генетической диагностике, особенно это необходимо делать семейным парам с неблагоприятным семейным анамнезом.
Хондродисплазии: классификация, нарушения, диагностика, лечение
Это группа наследственных заболеваний опорно-двигательного аппарата, которые часто проявляются карликовостью и нарушениями пропорций тела.
У некоторых больных рост и телосложение нормальные, но имеются характерные изменения глаз или расщелина неба, часто встречающиеся и у более тяжелых больных.

Нередко развиваются дегенеративные изменения суставов, поэтому легкие формы хондродисплазий у взрослых трудно отличить от генерализованного деформирующего остеоартроза.
Некоторые авторы предпочитают называть эту группу болезней скелетными дисплазиями, но термин «хондродисплазии» более распространен.
Классификация
Выделено более 150 типов хондродисплазий, составляющих 8 основных групп.
Группа ахондроплазии
- Ахондроплазия
- Гипохондроплазия
- Танатофорная дисплазия
Спондилоэпифизарные дисплазии
- Ахондрогенез, тип IA (тип Хьюстона—Харриса)
- Ахондрогенез, тип IB (тип Фраккаро)
- Ахондрогенез, тип II (тип Пантера—Салдино)
- Гипохондрогенез
- Врожденная спондилоэпифизарная дисплазия Спондилоэпиметафизарная дисплазия, тип Страдвика (синдром Страдвика)
- Синдром Стиклера
- Дисплазия Книста
Точечные хондродисплазии
- Ризомелическая точечная хондродисплазия
- Точечная хондродисплазия Конради—Хюнермана (аутосомно-доминантная)
- Х-сцепленная доминантная точечная хондродисплазия
- Х-сцепленная рецессивная точечная хондродисплазия
Синдромы коротких ребер
- Хондроэктодермальная дисплазия (синдром Эллиса—ван Кревельда)
- Асфиктическая дисплазия грудной клетки
- Синдром коротких ребер, тип I (синдром коротких ребер — полидактилии, тип Салдино—Нунан)
- Синдром коротких ребер, тип II (синдром коротких ребер — полидактилии, тип Маевского)
Метатропные дисплазии
- Метатропная дисплазия
- Легкие формы метатропной дисплазии
Метафизарные хондродисплазии
- Метафизарная хондродисплазия, тип Янсена
- Метафизарная хондродисплазия, тип Шмида
- Метафизарная хондродисплазия, тип Мак-Кьюосика
Брахиолмия
- Брахиолмия I типа
- Брахиолмия II типа
- Брахиолмия III типа
Периферические дизостозы
- Периферический дизостоз
- Акродизостоз
- Трихо-рино-фалангеальный синдром, тип I
- Трихо-рино-фалангеальный синдром, тип II (синдром Лангера— Гидиона)
Выделяют хондродисплазии, приводящие к смерти (танатофорные), вызывающие искривление костей (диастрофические), поражающие метафизы (метафизарные), поражающие эпифизы (эпифизарные) и другие.
Некоторые хондродисплазии названы по имени первого или наиболее подробно описанного больного.
Тяжелые формы хондродисплазий сопровождаются выраженной деформацией большинства хрящевых структур и поражением глаз.
Легкие формы классифицировать труднее. Основные симптомы хондродисплазий — катаракта, дегенерация стекловидного тела, отслойка сетчатки, высокий лоб, гипоплазия лицевых костей, расщелина неба, короткие тонкие конечности и выраженная деформация эпифизов, метафизов и суставных поверхностей.
Распространенность
Хотя точная распространенность большинства хондродисплазий неизвестна, возможно, что это одни из самых частых наследственных болезней соединительной ткани: распространенность синдрома Стиклера типа I (артроофтальмопатии), например, достигает 1:10 000.
Молекулярные нарушения
Первые мутации при хондродисплазиях были выявлены в гене COL2A1, кодирующем коллаген II типа — основного белка хрящевой ткани. Всего в этом гене выявлено более 40 мутаций, вызывающих различные хондродисплазии.
Мутации гена COL2A1 обнаружены примерно у 20% больных с тяжелыми и среднетяжелыми хондродисплазиями и у 2% больных с ранним генерализованным деформирующим остеоартрозом. Сходная клиническая картина может быть вызвана также мутациями других генов: трех других типов коллагена, входящих в состав хряща; факторов роста; рецепторов факторов роста; факторов транскрипции.
Число описанных мутаций не отражает реальной генетической гетерогенности той или иной болезни: оно зависит от сложности структуры гена, технических трудностей, доступности больших семей для генодиагностики и интенсивности исследовательской работы. Очевидно, будут выявлены новые мутации.
Мутации гена COL2A1 приводят к дефектам проколлагена II типа. Большинство из 40 известных таких мутаций вызывают тяжелые хондродисплазии, такие, как ахондрогенез типа II (тип Лангера—Салдино) и дисплазия Книста. Однако мутации этого гена обнаружены и в некоторых семьях, члены которых в детстве были здоровы, но в зрелом возрасте у них появились симптомы деформирующего остеоартроза (боль, скованность и дегенеративные изменения суставов).
Наследственные дефекты проколлагена II типа сходны с дефектами проколлагенов I и III типов, и при них так же трудно установить соответствие генотипа и фенотипа. Тем не менее известно, что мутации, вызывающие образование терминирующего кодона и синтез укороченного белка, приводят к синдрому Стиклера типа I (артроофтальмопатии).
При деформирующем остеоартрозе с легкой формой хондродисплазии обнаружены мутации, приводящие к замене аргинина на цистеин в положении Y повторяющейся последовательности -Гли-X-Y-.
При метафизарной хондродисплазии (тип Шмида), которая проявляется низкорослостью, Х-образным искривлением ног, искривлением метафизов и утиной походкой, обнаружены дефекты коллагена X типа, короткие молекулы которого синтезируются в основном хондроцитами II типа и образуют сеть в межклеточном веществе хряща. Некоторые формы синдрома Стиклера (тип III, то есть без поражений глаз) вызваны дефектами α2(Х1)-цепи коллагена XI типа, малораспространенного коллагена, входящего в состав хряща и других тканей.
У большинства больных ахондроплазией (это самая частая причина непропорциональной низкорослости, сопровождающейся макроцефалией и дисплазией метафизов длинных трубчатых костей) обнаружены мутации гена FGFR3, кодирующего рецептор фактора роста фибробластов.
Замена лишь одного нуклеотида, вызывающая замену глицина на аргинин в положении 380 молекулы рецептора, имеется почти у 90% больных. Обычно это новые мутации, так что эта точечная мутация — одна из наиболее часто возникающих мутаций в геноме человека. Она приводит к конститутивной (не зависящей от лиганда) активации рецептора и нарушению развития хряща.
При более тяжелых болезнях, таких, как гипохондроплазия и танатофорная карликовость, а также в некоторых семьях с краниосиностозом мутации затрагивают другие участки гена FGFR3. Однако у большинства больных с краниосиностозом обнаруживают мутации родственного гена FGFR2.
При множественной эпифизарной дисплазии и псевдоахондроплазии, родственных болезнях, проявляющихся укорочением конечностей и деформирующим остеоартрозом, обнаружены мутации гена СОМР, кодирующего олигомерный белок хрящевого матрикса. Однако в одной семье с эпифизарной хондродисплазией обнаружена мутация гена COL9A1, кодирующего а2(1Х)-цепь коллагена IX типа.
Диагностика
Диагноз при тяжелых формах хондродисплазий ставят на основании характерной внешности, рентгенологических данных, гистологических изменений и течения болезни. Иногда, хотя и реже, чем при несовершенном остеогенезе, оказывается возможной пренатальная диагностика с помощью УЗИ. Скоро станут доступными методы выявления мутаций гена COL2A1.
Лечение хондродисплазий не разработано. Проводят симптоматическое лечение деформирующего остеоартроза и других проявлений болезни.
Многим больным проводят протезирование суставов и устранение расщелины неба.
Больных должен регулярно осматривать офтальмолог, чтобы своевременно обнаружить развитие катаракты и провести лазерную коагуляцию при отслойке сетчатки.
Больным советуют следить за весом и избегать контактных видов спорта.
При низкорослости очень важна психологическая помощь, с этой целью во многих странах создают группы поддержки.
-0 Комментарий-